Research
Our long-term goal is gain insights into the fundamental mechanisms that determine gene expression changes during cell fate transitions. During differentiation and development, cells change their gene expression program and form specific cell types that become increasingly resistant to cell fate changes. Thus, differentiated cell states are remarkably stable in vivo. However, they can be reprogrammed experimentally to pluripotency, or into other lineages, by overexpression of only a few transcription factors. In our laboratory, we therefore use differentiation and reprogramming processes as powerful in vitro models for studying the molecular mechanisms underlying cell fate changes. We are trying to understand how histone and DNA modification patterns are established (and erased) during cell fate changes, and how in turn chromatin states determine developmental potency. We often examine the epigenetic changes underlying cell fate changes through the lens of the X-chromosome inactivation. This remarkable process induces silencing and heterochromatin formation on an entire chromosome through the function of the long-noncoding RNA Xist, giving us a glimpse of the role of long-noncoding RNAs and a look at how heterochromatin forms during differentiation and is reset during reprogramming. We are also interested in uncovering how the genome is folded in the nucleus to fulfill its functions, how this organization changes during reprogramming and differentiation, and how genome folding interacts with transcriptional networks and lncRNAs. The processes we examine are centered on the pluripotent state with its remarkable property to be able to produce every cells type of the body.
ENGINEERING CELL FATE CHANGE - THE REPROGRAMMING PROCESS
We are well known for our pioneering work on somatic cell reprogramming to pluripotency, including the description of fully reprogrammed mouse induced pluripotent stem cells (iPSCs) ( Maherali et al 2007 ), establishment of human iPSCs ( Lowry et al, 2008 ), the characterization of the epigenetic state of mouse and human iPSCs ( Maherali et al 2007); Chin et al 2009 ), the comparison of chromatin and gene expression states between embryonic stem cells (ESCs) and iPSCs ( Chin et al 2009 and 2010 ), the definition of requirements of the reprogramming process ( Ho et al, 2013; Sridharan et al 2013 ), the identification of reprogramming steps ( Pasque et al, 2014, ), the elucidation of reprogramming factor binding ( Sridharan et al 2009 ), and the use of the inactive X chromosome to reveal insights into the chromatin reorganization during the reprogramming of human and mouse cells ( Maherali et al 2007, Tchieu et al 2010 ). The ability to generate all cell types of the body has thrust ESCs and iPSCs into the spotlight as a panacea for regenerative medicine. With the generation of human iPSCs it became feasible to generate pluripotent cells from any person by reprogramming, which has ushered in an era of personalized medicine and “disease-in-a-dish” modeling, which has encouraged various collaborations with the Lowry lab at UCLA (see collaborations).
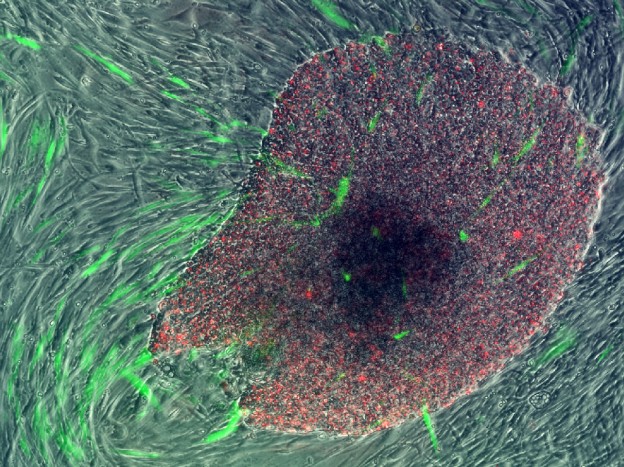
One of our first human iPSC colonies
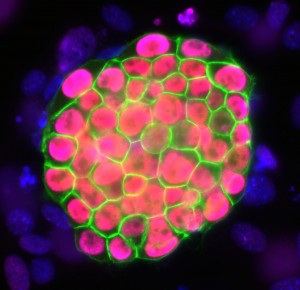
mouse iPSC colony
Current research in the Plath is aimed at systematically defining how the reprogramming transcription factors reset the identity of differentiated cells to pluripotency, and determining the interplay of the reprogramming factors with somatic chromatin states and changing signaling environments, to understand how cell fate decisions can be modulated. Recently, we have shown that reprogramming cells progress through specific stages that can be defined based on global and inactive X chromatin states (Pasque et al, 2014), raising the question of what enables transitions from one to the next stage in decreasing numbers of cells, yielding pluripotency in only few cells at the end of the process. One of our goals within the reprogramming problem therefore is to determine the specific sequences of dynamic events (at the gene expression and chromatin level) that occur in the reprogramming cells as they are moving along to pluripotency. By obtaining a direct view of how cells transition to pluripotency, we will provide a quantum leap in our understanding of the molecular basis of cell fate change processes.
For us, the projects on reprogramming of somatic cells to iPSCs are critical to the overall mission of our laboratory for three reasons: First, the reprogramming process serves as a unique tool for the mechanistic dissection of the role of chromatin, transcription factors, signaling pathways, 3D genome organization, and long-noncoding RNAs during cell fate change processes. Second, the reprogramming process allows us to develop and test novel approaches to reveal the molecular basis of cell fate transitions. Third, by understanding why specific transcription factors are particularly efficient in inducing pluripotency while others lack this ability, we can better define the sets of transcription factors best suited for engineering cell fate.
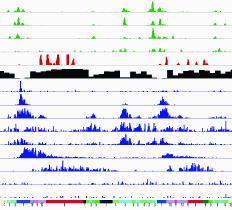
Mapping reprogramming
X-INACTIVATION - SILENCING OF AN ENTIRE CHROMOSOME BY A LONG-NONCODING RNA
Our lab is committed to dissect the molecular basis of the X chromosome inactivation process. X chromosome inactivation refers to the silencing of one of the two X chromosomes in female placental mammals and serves to equalize the dose of X-linked genes between XX females and XY males. Xist (X-inactive specific transcript), a 17kb long noncoding RNA (lncRNA) in its spliced form and encoded on the X, is the master regulator of XCI. Xist is one of the first lncRNAs that was identified and specifically marks the inactive X chromosome (Xi) in female differentiated cells. It has been show by the field that deletion of Xist prevents the inactivation on the X chromosome in cis, and its forced expression is sufficient to induce chromosome-wide silencing in cis, even when placed on autosomes. Based on these findings, the widely accepted model is that upregulation of Xist on one of the two X chromosomes and its spreading in cis induce X-inactivation.
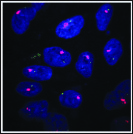
Xist RNA (pink) detected by FISH
X-chromosome inactivation occurs when the blastocyst implants and can be recapitulated with differentiating female mouse ESCs, which provides a versatile experimental system to uncover the underlying molecular mechanisms. In undifferentiated mouse ESCs, both X chromosomes are active and Xist is largely repressed. At the onset of differentiation, Xist becomes strongly expressed on the future Xi. Xist then quickly spreads along the length of X chromosome, forming a ‘coat’ and inducing gene silencing. Thus, the X-chromosome inactivation process leads to the silencing of an entire chromosome specifically when cells differentiate, which provides a fascinating example of gene regulation closely linked to the loss of pluripotency.
We already know several functions of Xist, including the spreading in cis from its site of transcription to physically cover the entire X chromosome, the exclusion of RNA Polymerase II, the accumulation of repressive chromatin regulators including the polycomb repressive complex 2 (PRC2), and the induction of a chromosome-wide compaction, demonstrating the remarkable versatility of this lncRNA. However, the mechanisms by which Xist carries out these functions remain poorly understood. In particular, with few exceptions, we still do not know domains of Xist or interacting proteins that mediate these various functions.
Our questions: In our work, we therefore aim to decipher how Xist becomes expressed appropriately, how Xist RNA localizes along the entire X chromosome, how the RNA interacts with protein complexes, and how, through these interactions, Xist RNA controls chromosome conformation, heterochromatin formation, and gene silencing across the X chromosome. Moreover, upon the establishment of the inactive X, the silent state is faithfully maintained through the lifetime of the organism, and we are interested in understanding the mechanisms that contribute to that extreme stability of the Xi through cell division (Minkovsky et al 2014). Mammalian genomes encode thousands of long-noncoding (lnc) RNAs, many of which play key roles in the regulation of gene expression. We believe that Xist RNA is an ideal model to investigate these mechanisms of lncRNA function.
The research on X-chromosome inactivation in the Plath lab also extends to the reprogramming process. We demonstrated that X-inactivation and the reactivation of the inactive X are distinctly different between mouse and human reprogramming models (Maherali et al 2007, Tchieu et al 2010), which led to the identification of an epigenetic instability of human iPSCs/ESCs that affects the utility of these cells and challenges conventional wisdom about X-inactivation (Tchieu et al 2010, unpublished). Based on this work, we have become interested in studying the regulation of X-inactivation in human development. We hope that the work on the X-inactivation model in both the human and mouse systems will promote our understanding of how epigenetic processes such as X-inactivation can be adopted in evolution and influence developmental potency.
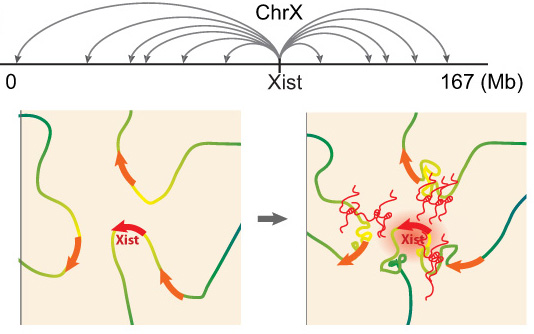
Xist exploits 3D genome organization to spread across the X
Example of a recent project: In collaboration with Mitch Guttman (Caltech, see collaborations), we recently explored Xist RNA localization at high resolution using the RAP-seq method developed by Mitch. Together, we discovered that Xist spreads quickly from its site of transcription to about 30 spatially proximal, but linearly distal, regions of the X chromosome. This finding suggests that extensive (pre-existing) long-range chromatin interactions of the genomic Xist locus are essential for Xist to spread, and thereby for the X-inactivation process. This is the first model to explain how Xist-RNA spreads along the X-chromosome (Engreitz et al 2013). Interestingly, at its distal entry sites, we found that Xist initially more effectively accumulates at non-genic regions and silent genes and only a little later moves efficiently over active genes. Intriguingly, based on published RNA FISH studies, active genes are initially located on the edge of the Xist cloud, but are in the central Xist compartment upon silencing. Therefore, we hypothesize that Xist mediates changes in 3D organization of the X chromosome as it spreads over active genes to promote silencing. We also propose that by repositioning previously unbound regions into its growing compartment, Xist effectively pulls new regions of chromatin closer to its transcription locus, thus allowing the RNA to spread to new sites by proximity transfer, ultimately localizing along the entire X-chromosome. The coordinated interplay between lncRNA localization and chromosome conformation may have broader implications beyond Xist. Other lncRNAs may similarly take advantage of chromosome conformation to identify target sites in close spatial proximity, which could even reside on other chromosomes. This search strategy capitalizes on the unique ability of a lncRNA to act while tethered to its transcription locus, in contrast to an mRNA which requires export and translation to carry out its function. Because chromosome conformation is nonrandom, a proximity-guided search strategy might explain how low-abundance lncRNAs can reliably identify their genomic targets. Upon binding these targets, lncRNAs may in turn alter chromosome conformation through their interactions with various chromatin regulatory complexes. We are now pushing towards a better understanding of the molecular mechanisms underlying Xist’s localization.
REGULATION OF GENE EXPRESSION AND CHROMATIN STATES IN 3D
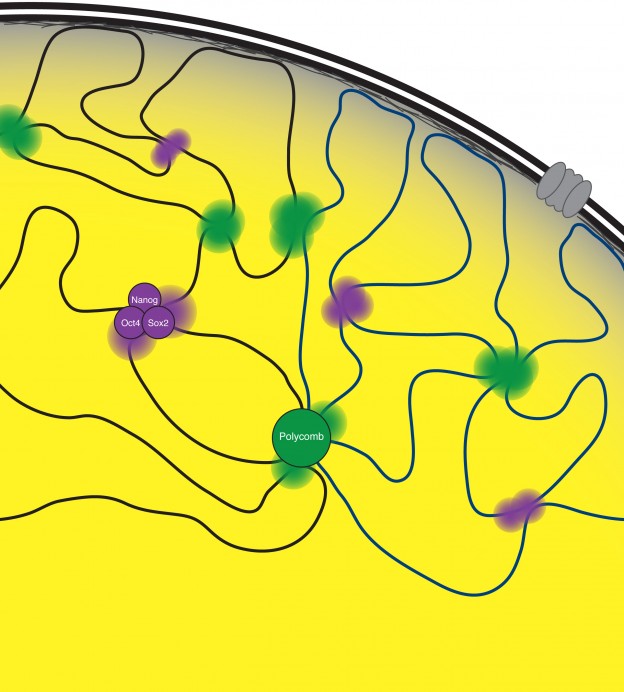
3D chromatin organization in ESCs
The DNA of eukaryotic cells is organized in a non-random fashion that allows for the accessibility of the gene regulatory machinery and for transcriptional and epigenetic changes that accompany cell fate changes. We discovered that transcription factors and chromatin regulators play a key role in bringing together stretches of DNA from distant sites in the genome, providing a mechanism for how co-regulated regions co-localize in 3D in the nucleus (Denholtz et al 2013). We are now developing models that comprehensively describe this spatial organization of the genome and the underlying molecular mechanisms; explore how this spatial organization of the genome contributes to the control of gene expression, and how changes in genome organization control cell fate transitions, cellular function, and disease processes. A second intriguing aspect of genome organization was revealed by our recent work on the mechanism of X-inactivation described above, providing an intricate link between genome organization and lncRNAs. For us, the X-inactivation paradigm therefore provides a unique opportunity to gain insights into the molecular mechanisms of genome organization, its dynamic regulation and relevance for gene and chromatin regulation.